Hypertrophic Cardiomyopathy (HCM)
What is HCM?

- Hypertrophic cardiomyopathy (HCM) is a heart condition where the muscle of the heart becomes thickened over time. Usually, this thickening occurs in the muscle of the main pumping chamber of the heart, the left ventricle.1
- In HCM the heart muscle contracts (or squeezes) harder than it needs to, building up more muscle and becoming thick and stiff. Over time, this thickness prevents the pumping chambers of the heart from relaxing and expanding to fill with blood.1
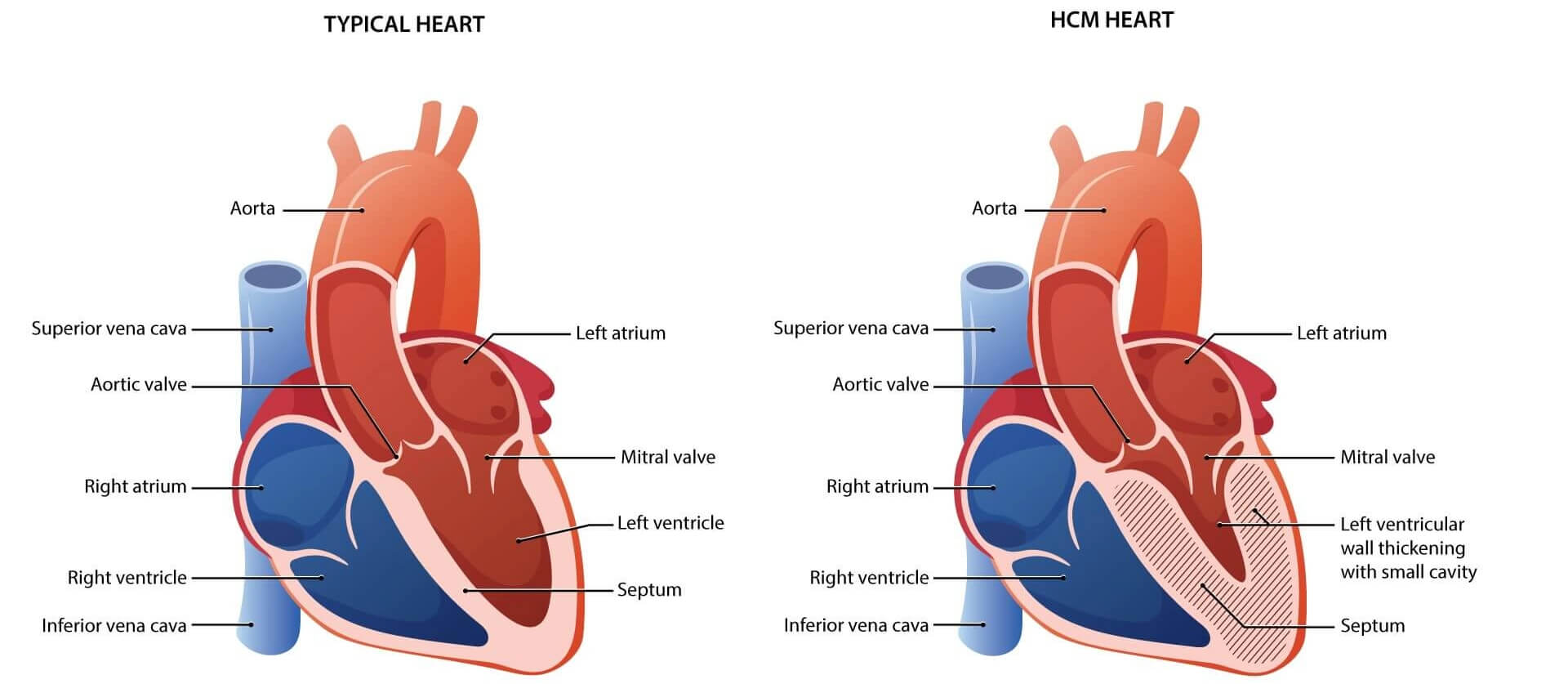
- As a result of the heart muscle not working as expected, people with HCM can develop a variety of symptoms such as shortness of breath, fainting, fatigue and chest pain.1
- Symptoms can be so serious that they frequently get in the way of doing everyday activities.
- HCM can also lead to more urgent symptoms like irregular heartbeats, heart failure or sudden death.
- HCM is a chronic, progressive disease, meaning that once someone becomes symptomatic, their symptoms usually do not go away, and over time their condition is likely to worsen.
It is estimated that one in every 500 people have HCM, or approximately 600,000 people in the U.S.2 Most people with HCM have not been diagnosed.

What causes HCM?
Researchers are still studying all the causes of HCM but know that the majority of HCM cases (more than 60%) are caused by genetic mutations, or changes or variations, in certain heart muscle genes.3 Mutations in a gene can affect that gene’s ability to make a protein the way it should or can affect whether that protein is made at all. When not enough protein is made, or not made at all, the function of the heart muscle cells is impaired.
Mutations in Genes Cause HCM4

The most common cause of HCM is a mutation in a gene called MYBPC3, which stands for myosin-binding protein C.5
- The MYBPC3 mutation is found in about 20% of people diagnosed with genetic HCM.6
- Changes in the MYBPC3 gene can prevent the heart muscle cells from making enough of a protein called myosin-binding protein C.
- This protein is needed for the cells of the heart muscle to function as expected. Without enough myosin binding protein C, the heart contracts harder than it needs to, leading to HCM.
- People with HCM whose condition is caused by mutations in the MYBPC3 gene may have more severe disease, where they develop symptoms at a younger age and/or their symptoms worsen more quickly than others.
Inheritance Patterns of HCM8
HCM is the most common inherited form of heart disease.7 Genetic mutations for HCM are usually inherited (passed down) from one parent who carries the genetic mutation.8
- A child can develop HCM if they receive one copy of the MYBPC3 mutation from one parent (called an autosomal dominant inheritance pattern).
- In rare cases, HCM has an autosomal recessive pattern of inheritance, in which two copies of the MYBPC3 mutation, one from each parent, can cause the condition.
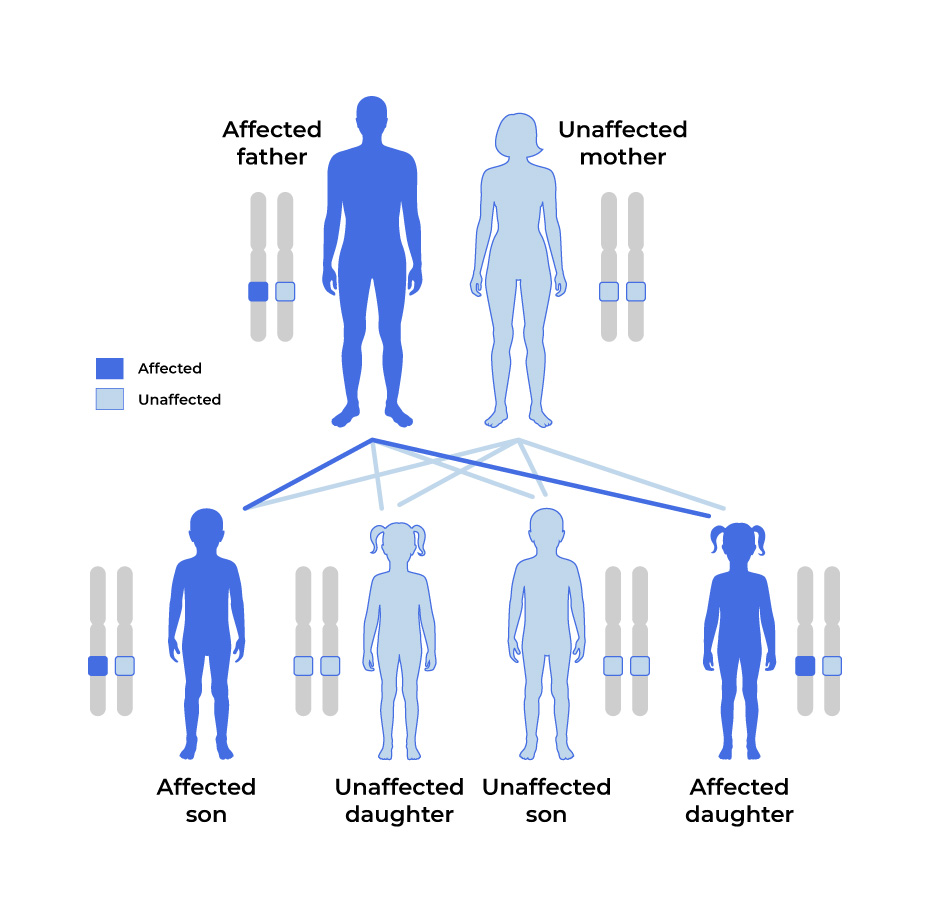
Autosomal Dominant
In most cases of HCM, the genetic mutation is carried by one parent and passed down to children.
With an autosomal dominant pattern, there is a 50% chance of passing the mutation down to a child.
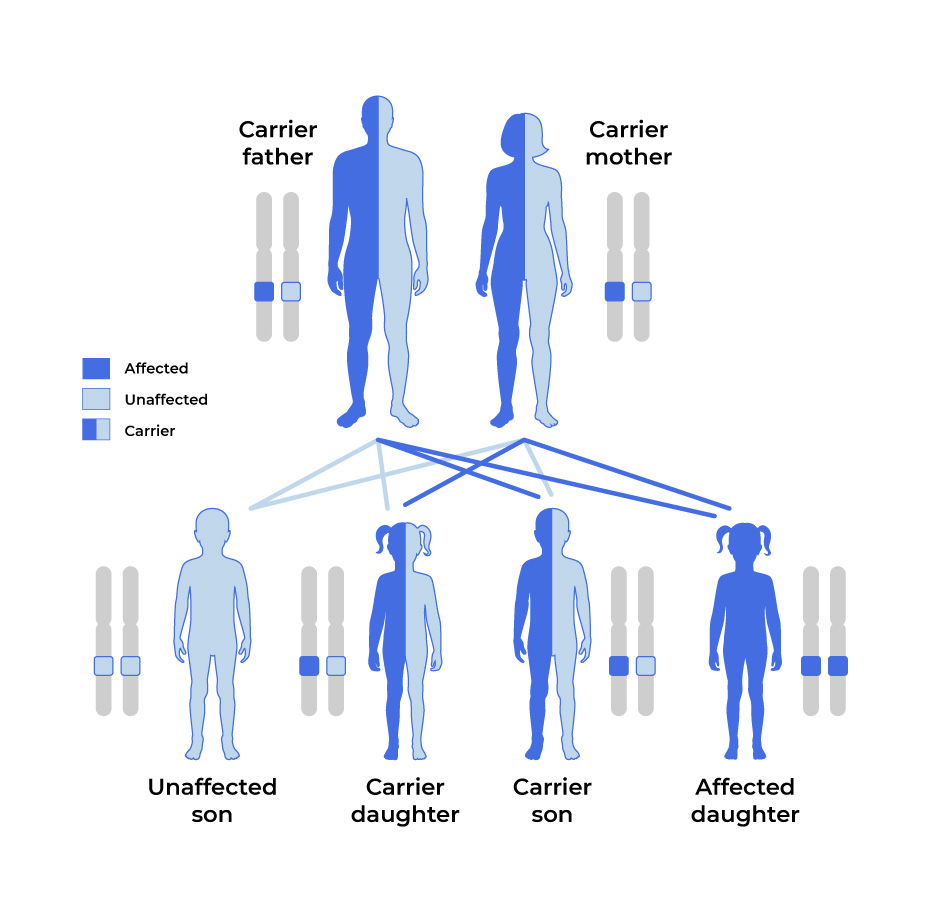
Autosomal Recessive
In more rare circumstances, neither parent is affected by HCM but carries the mutation.
With an autosomal recessive pattern, there is a 25% chance that both parents pass the mutation down to a child.
People who have been diagnosed with HCM but who don’t yet know their specific mutation should talk to their cardiologist, primary care physician, or a genetic counselor about genetic testing. Click here for more information about genetic testing.
What are the symptoms of HCM?
Some people have described HCM as being like their heart can go into a state of “fight or flight” — even when they are relaxed and at rest.

The most common symptoms of HCM include:1
- Chest pain
- Shortness of breath
- Fatigue (tiredness)
- Fainting
- Dizziness
- Rapid or irregular heartbeat
- Swelling in the ankles, feet, legs, and abdomen
How is HCM diagnosed?
HCM can sometimes be difficult to diagnose because many of the symptoms that first appear are common to other diseases.
- HCM may be diagnosed based on the onset of symptoms, family screening, or the discovery of an abnormal electrocardiogram (ECG) pattern.
- For a definitive diagnosis, a cardiologist will use an imaging tool, known as an echocardiogram (echo) to measure the thickness of the heart muscle.
Genetic testing is also an essential part of the diagnosis and management for people with HCM and their family members. According to the American Heart Association, the American College of Cardiology, and the European Society of Cardiology, genetic testing is recommended if a person, or their family members, is affected by HCM.9,10
- Results of genetic testing may reveal that a person with no HCM symptoms has genetic mutations known to cause HCM.
- If a person knows that one of their relatives has HCM, they should tell their doctor.
- In either case, it may be helpful to consult with a specialized cardiologist or a comprehensive HCM center. Specialists or comprehensive care centers can
- Help with genetic testing and genetic counseling,
- Recommend a plan to monitor a person’s heart,
- Make suggestions on how to best take care of themselves, and
- Have knowledge of or access to the latest treatments and clinical trials.
What is the role of a genetic counselor?
A genetic counselor is a healthcare professional that a person may see as part of the genetic testing process. Genetic counselors can help explain:
- The genetic testing process and possible outcomes
- How a condition is inherited
- Individual test results
- The chance of passing a gene mutation on to a child
How can I find a genetic counselor?
Ask your doctor or find more information at the National Society of Genetic Counselors.
Expand for References
1. What is hypertrophic cardiomyopathy? American Heart Association; 2022. https://www.heart.org/-/media/Files/Health-Topics/Answers-by-Heart/What-is-HCM.pdf. Accessed April 15, 2024. 2. Semsarian C, Ingles J, Maron MS, et al. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol. 2015;65(12):1249-1254. https://doi.org/10.1016/j.jacc.2015.01.019. 3. Marian AJ, Braunwald E. Hypertrophic cardiomyopathy: genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ Res. 2017;121(7):749-770. https://doi.org/10.1161/CIRCRESAHA.117.311059. 4. Crilley JG, et al. Hypertrophic cardiomyopathy due to sarcomeric gene mutations is characterized by impaired energy metabolism irrespective of the degree of hypertrophy. J Am Coll Cardiol. 2003;41:1776-1782. https://doi.org/10.1016/s0735-1097(02)03009-7. 5. van Velzen HG, Schinkel AFL, Oldenburg RA, et al. clinical characteristics and long-term outcome of hypertrophic cardiomyopathy in individuals with a MYBPC3 (Myosin-Binding Protein C) founder mutation. Circ Cardiovasc Genet. 2017;10(4):e001660. https://doi.org/10.1161/CIRCGENETICS.116.001660. 6. Sedaghat-Hamedani F, Kayvanpour E, Tugrul OF, et al. Clinical outcomes associated with sarcomere mutations in hypertrophic cardiomyopathy: a meta-analysis on 7675 individuals. Clin Res Cardiol. 2018;107(1):30–41. https://doi.org/10.1007/s00392-017-1155-5. 7. Czepluch FS, Wollnik B, Hasenfuß G. Genetic determinants of heart failure: facts and numbers. ESC Heart Fail. 2018;5(3):211-217. https://doi.org/10.1002/ehf2.12267. 8. Overview of Inheritance. American Heart Association. https://www.heart.org/-/media/Files/Health-Topics/Cardiomyopathy/Cardiomyopathy-Inheritance-UCM_312228.pdf. Accessed April 15, 2024. 9. Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: A report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2020;142(25):e558-e631. https://doi.org/10.1161/CIR.0000000000000937. 10. Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 2014;35(39):2733-2779). https://doi.org/10.1093/eurheartj/ehu284.